



![]()

| Project coordinator (PC):Estivill, Xavier(CRG) | Grant Agreement Nº:HEALTH-2010-261123 | |
| Call: 7thFramework Programme (2010) – HEALTH | Scheme: Consortium Action | |
| Total budget: 2,000.000,00€ | Start date: 01/10/2010 | Term: 3 years |
| Web site: http://www.geuvadis.org/ | ||
| Project summary: high-throughput next-generation DNA sequencing technologies allow investigators to sequence entire human genomes at an affordable price and within a short time frame . The correct interpretation, storage, and dissemination of the large amount of produced genomics data generate major challenges. Tackling these challenges requires extensive exchange of data, information and knowledge between medical scientists, sequencing centres, bioinformatics networks and industry at the European level. The GEUVADIS (Genetic European variation in disease) Consortium aims at developing standards in quality control and assessment of sequence data, models for data storage, exchange and access, as well as standards for the handling, analysis and interpretation of sequencing data and other functional genomics datasets, standards for the biological and medical interpretation of sequence data and in particular rare variants for monogenic and common disorders, and finally standards for the ethics of phenotype prediction from sequence variation. The partners are all involved in international sequencing initiatives (1000 GP, ICGC), EU and other international projects (ENGAGE, GEN2PHEN, ENCODE, TECHGENE …), biobanking activities (BBMRI), data sharing initiatives (ELIXIR), and the European Sequencing and Genotyping Infrastructure (ESGI), ensuring tight collaborations. The Consortium will undertake pilot sequencing projects on selected samples from three medical fields (cardiovascular, neurological and metabolic), using RNA (RNASeq) and DNA (exonSeq) sequencing. The analysis of such samples will allow the consortium to set up standards in operating procedures and biological/medical interpretation of sequence data in relation to clinical phenotypes. The consortium will bring together the knowledge and resources on medical genome sequencing at a European level and allow researchers to develop and test new hypotheses on the genetic basis of disease.
Key Words: high- throughput sequencing technologies, DNA, human genome, data exhange, interpretation storage, models, bioinformatics, rare variants, (clinical) phenotype predictions, genetic bases of disease, operation procedure standards; hypotheses, knowledge, ethics. |
||
LIST OF BENEFICIARIES
| Partner | Name | Acronym | Type/ Category |
| 1 | Centre for Genomic Regulation (ESP) | CRG | Public Research Institute |
| 2 | University of Genevè(SWZ) | UNIGE | Public/ higher education & research |
| 3 | Helmholtz Zentrum München (GER) | HMGU | Public/ higher education & research |
| 4 | Wellcome Trust Sanger Institute (UK) | WTSI | Public / Healthcare & Research |
| 5 | Centre National de la Recherche Génomique (FRA) | CNG | Public /Research Institute |
| 6 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften (GER) | MPI-MG | Public Research Institute |
| 7 | Centro Nacional Avanzado de Genómica (ESP) | CNAG-PCB | Public Research Institute |
| 8 | Uppsala University (SWE) | UU | Public/ higher education & research |
| 9 | Christian-Albrechts-University Kiel (GER) | CAU | Public/ higher education & research |
| 10 | Radboud University Nijmegen Medical Centre (NE) | RUN-MC | Public/ higher education & research |
| 11 | Academisch Ziekenhuis Leiden – Leids Universitair Medisch Centrum (NE) | LUMC | Public/ higher education & research |
| 12 | University of Santiago de Compostela (ESP) | USC | Public/ higher education & research |
| 13 | European Molecular Biology Organization / European Bioinformatics Institute (UK) | EMBL/EBI | Public Research Institute |
| 14 | Institut National de la Santé et de la Recherche Médicale (FRA) | INSERM | Public / Healthcare & Research |
| 15 | Applied Biosystems Deutschland GmbH (GER) | AB | Private/ Industry |
| 16 | Illumina Cambridge Limited (UK) | Illumina | Private/ Industry |
| 17 | Johns Hopkins University School of Medicine (USA) | JHU/OMIM | Private / Healthcare & Research |
TEAM (*)
| Surname | Name | Title/ Position | Entity |
| Estivill | Xavier | MD, PhD / PC | CRG |
| Guigo | Roderic | Bioinformatics & Genomics Program, PhD / PI | |
| Dermitzakis | Emmanouil | Professor of Genetics, PhD / PI | UNIGE |
| Antonarakis | Stylianos | Professor& Chairman – Genetic Medicine, MD | |
| Meitinger | Thomas | Director of HMGU, Professor at TUM, PhD / PI | HMGU |
| Strom | Tim M. | Sequencing Unit Head, PhD | |
| Palotie | Aarno | Head of Medical Sequencing, PhD / PI | WTSI |
| Lathrop | Mark | CEA-IG / PI | CNG |
| Lehrach | Hans | Director at the MPI-MG, PhD / PI | MPI-MG |
| Sudbrak | Ralf | 1000 Genome Project Coordinator, PhD | |
| Gut | Ivo | Director of CNAG, PhD/ PI | PCB-CNAG |
| Syvänen | Ann-Christine | Professor of Molecular Medicine, PhD / PI | UU |
| Lindblad-Toh | Kerstin | Guest Professor in Comparative Genomics, PhD | |
| Schreiber | Stephan | Director of the ICMB,Head of Internal Medicine Department,MD/ PI | CAU |
| Rosenstiel | Philip | Professor of Molecular Medicine,MD | |
| Veltman | Joris | Head of Genomic Disorders Group, PhD/ PI | RUN-MC |
| Brunner | Han | Head of Human GeneticsDepartment, PhD | |
| van Ommen | Geert J. | MD, PhD / PI | LUMC |
| Carracedo | Angel | Professor of Legal Medicine, Director of FPGMX,MD, PhD / PI | USC-FPGMX |
| Brazma | Alvis | MD, PhD / PI | EMBL/EBI |
| Flicek | Paul | Vertebrate Genomics Team Leader, PhD | |
| Cambon-Thomsen | Anne | Research Director,MD, PhD / PI | INSERM |
| Mangion | Jonathan | Sr. Manager Bioinformatic Suppport,PhD/ PI | AB |
| Bentley | David | Vice President, Chief Scientist and Head of Genomic Applications, PhD/ PI | Illumina Inc. |
| Hamosh | Ada | Professor of Pediatricsand IGM, Scientific Director at OMIM,MD, PhD / PI | JHU/OMIM |
(*) For the sake of brevity and conciseness, only PIs and relevant member(s) from the different entities involved have been listed.PC: Project CoordinatorPI: Principal Investigator.
Background to the project/main objectives. Work plan and cronogram.
The latest high-throughput next-generation sequencing technologies allow investigators to sequence entire human genomes and transcriptomes at an affordable price and within a short time frame. An increasing number of research centres in Europe have access to these technologies, in-house or through regional, national and international infrastructures. Storing, disseminating and analysing the large amount of data produced generate major challenges. Tackling these challenges requires extensive exchange of data, information and knowledge between sequencing centres, bioinformatics networks, the medical research community and the industry at the European level. The GEUVADIS (Genetic EUropeanVAriation in DISease) Consortium had four main aims:
- Develop standards in quality control (QC) and assessment of sequence data (WP2).
- Develop models for sequencing data storage, access and exchange (WP3).
- Develop standards for the handling, analysis and interpretation of sequencing data from DNA and RNA (WP4 and 5).
- Develop guidelines on the handling of ethical, legal and social implications (ELSI) of phenotype prediction from sequence variation (WP6).
The consortium through the GEUVADIS website (GEUVADIS.com) will provide a framework for exchange of expertise and data not only between consortium members but with any other European centre working in genomic medicine. It will bring together the knowledge and resources on medical genome sequencing at a European level and allow researchers to develop and test new hypotheses on the genetic basis of disease. This will allow the European research community to contribute significantly to the development of genomics-based applications for diagnostics and therapeutics.
In order to achieve our goals, the following work plan and cronogram were put in place:
| WP nr. | Title/ Name of Working Package | Effort
(months) |
Lead participant |
| WP1 | Coordination and Communication Office | 36 | P1 (CRG) |
| Communication and coordination | |||
| Events | |||
| WP2 | Quality Control of Sequence Data | 14 | P2 (UNIGE) |
| T 2.1. | Survey | ||
| T 2.2 | Best practice | ||
| T 2.3 | Recommendation for the community | ||
| WP3 | Data Storage, Access and Exchange | 36 | P13
(EMBL-EBI) |
| Recommendations | |||
| Analysis components | |||
| Data deposition | |||
| Data visualization | |||
| EVA (European Variation Archive) | |||
| WP4 | Handling, Analysis and Interpretation of RNA-sequence data and other functional datasets | 24 | P2 (UNIGE) |
| Study design | |||
| Data analysis | |||
| Main scientific findings | |||
| WP5 | Biological and Medical Interpretation of Sequence Data for Rare Variants | 24 | P3 (HMGU) |
| Sequencing tools in Europe | |||
| Patient consent and data access | |||
| Sequencing consortia | |||
| GEEVS: the GEUVADIS European Exome Variant Server | |||
| WP6 | Ethical, Legal and Social Issues of Phenotype prediction from Sequence Variation | 36 | P14 (INSERM) |
| WP7 | Dissemination and Training | 18 | P11 |
Geuvadis project’s prior art and literature references.
Genomics is making faster progress than any other technology field in biomedicine. Advent of whole human genome sequencing for thousand of individuals will be a reality within the next few years, actually the cost of sequencing has drop to the point where a whole human genome is now available for about €60,000. Although the costs will further fall with the advent of new technologies, it seems clear that it cannot be afforded at the clinical practice.During the last years new sequencing technologies have increased the sequencing throughput and reduced cost by several orders of magnitude (1- 4), including tumour samples (5). However, the costs of whole-genome sequencing are still too high for sequencing a considerable number of samples for either research or clinical purposes. Therefore investigation of large cohorts will concentrate on exome and transcriptome sequencing.
There are/ were several sequencing platforms under development that are going to supersede the current array-based methods of genomic analysis. These can be classified into two categories, those using multiplex cyclic sequencing by synthesis, and those not using synthesis. Some of the most interesting ones using synthesis are being developed by Pacific Biosciences, and Ion Torrent Systems. Some of the most exciting non-synthesis-based sequencing companies are Oxford Nanopore Technologies, NABsys and Halcyon. These are likely to replace current NGS, dominated by Illumina, SOLiD and Roche. However, the replacement will take several years and lveryikely a large number of genomic medicine discoveries and applications will be made by the current NGS platforms yet. Genomics, as an enhanced approach to healthcare, could this way transform the quality of life worldwide.
In addition to improving the cost and speed of existing genomic scanning, sequencing advances could open up the way to the eventual characterization of the whole cell and its interactions through the sequencing of the transcriptome, the proteome, the metabolome, the microbiome and other biological features, including histone modification sequencing, DNA methylation, acetylation and phosphorylation. It seems quite likely that whole human genome sequencing would be a routine component of everyone health record available to both patients and physicians for predictive and preventive healthcare purposes.
Genomic data is growing at 10x per year and research labs are confronted with rapidly increasing demands in respect to data storage, processing, access and data transfer. The raw data of the human genome is 3GB and the full collection of files in use for one whole human genome may reach 6TB, including intensity files, SAM (Sequence Alignment Map format), BAM (binary version of SAM), and other files with coordinates, variations etc.
The investigators of the present proposal are/ werein an excellent position to build on the expertise and technology developed in the 1000GP and the ICGC, and to incorporate data from these projects because several centres of this Consortium Agreement (CA) participate in either the experimental part or in the bioinformatics analysis of these two global projects.
Sequencing technologydeveloped rapidly during the last few years and new developments are expected to arrive during the lifetime of this CA. Each of the 12 sequencing centresin this proposal has several of the main sequencing technologies running (Genome Analyzer IIx / IIlumina and SOLiD / Applied Biosystems). At the time of starting this CA, the capacity of Genome Analyzer IIx (IIlumina) and SOLiD (Applied Biosystems) will reach between 100Gb and 200Gb per run.
New genome enrichment technologies can be used in combination with NGS, to generate deep sequencing of specific genome regions (6). The new methods use array hybridisation or liquid hybridization to biotinylated probes that target the genomic regions to be sequenced, followed by affinity-capture of the probe-hybrid complexes using streptavidin-coated magnetic beads (7). In the present proposal, we will use oligonucleotide capture technique based on liquid phase hybridization to capture exomes (collection of exons of the genome) from DNA libraries prepared from sheared genomic DNA. Several of the centres in our CA are already using these methods to study the exome in samples from individuals affected by different rare disorders.
The new sequencing technologies further allow the interrogation of the transcriptome by sequencing cDNA derived from total RNA, defined as RNAseq(8). RNAseq provides quantitative and qualitative profiles of the transcriptome by assessing the amount of transcript produced and by defining the contribution of individual exons and transcripts to the RNA of a given tissue (9, 10). RNAseq data is able to provide access to the dynamic range of transcription also giving access to low abundance transcripts (11).
Analysis of variability in gene expression in lymhoblastoid cell lines has shown that gene expression phenotypes are heritable in family pedigrees (12- 14). Other studies attempted to map genetic variants affecting expression levels of a limited number of genes in cis and in trans (15, 16). These studies have been facilitated by the availability of the “HapMap Consortium” with data for more than 3 million SNPs in 270 individuals from four populations.
The use of RNA sequencing has been very successful in the identification of a likely “driver” mutation in the FOXL2 gene in adult-type granulosa-cell ovarian cancer (17, 18). By sequencing the transcriptome of tumor specimens, rather than by sequencing genomic DNA, the investigators scanned all transcribed sequences to identify non-synonymous mutations in the tumour genome. By sequencing mRNA rather than genomic DNA the changes that affect the cellular-splicing machinery joining exons to single transcripts can efficiently be detected.
For RNA sequencingsome limitations have to be considered, such as the identification of somatic mutations in genes with low levels of expression by RNASeqwhich might become highly costly. Also, truncating somatic mutations that lead to nonsense-mediated decay may be missed. mRNA of good quality to prepare libraries may not always be available from cohort samples.
References:
- Shendure, 2008, Nat Biotechnol, 26:1135.
- Wang J et al, 2008, Nature, 4 56:60.
- Bentley et al, 2008, Nature, 456:53.
- Wheeler et al, 2008, Nature, 452:872.
- Ley et al. 2008, Nature, 456:66.
- Summerer et al. 2009, Genomics, 94363-368.
- Gnirke et al. 2009, Nature Biotech, 27:182-189.
- Morozova et al. 2009, Annu Rev Genomics Hum Genet, 10:135-51.
- Mortazavi et al. 2008; Nat. Methods, 5:621.
- Sultan et al. 2008; Science, 322:1849.
- Marioni et al. 2008 Genome Research, 18:1509.
- Monks et al. 2004.
- Morley et al. 2004.
- Schadt et al. 2003.
- Stranger et al. 2005.
- Cheung et al. 2005.
- Shah et al. 2009, N Engl J Med, 360:2719.
Overview of the main achievements of the project.
Since the start of the project in October 2010, the Consortium has obtained high impact results in all these areas:
1: After a detailed assessment of how GEUVADIS laboratories manage QC of Exome and RNA sequencing processes, we concluded that although the procedures for sample quantification, target enrichment and sequencing library preparation were quite similar across laboratories, there were substantial differences in the subsequent data analysis pipelines with respect to filtering procedures, alignment of sequencing reads and variant calling. Secondly, collaboratively established best-practice in quality control and sequence analysis of mRNA and small RNA. We successfully applied these practices in a large GEUVADIS study (see point 4 below).
2: We set up and implemented a data access and exchange strategy for the GEUVADIS projectRNA sequencing data. The data access scheme for the GEUVADIS RNA sequencing experiment issummarised in the scheme availableat http://www.geuvadis.org/web/geuvadis/RNAseq-project#Data_Access. Secondly, NGS data storage and exchange recommendations were developed and reported: Minimum Information about the Next generation Sequencing Experiment (MINSEQE) reporting standard, MAGE-TAB submission format for NGS data, and NGS terms in EFO ontology. Analysis components lists have been created and tested internally at EBI and externally by other GEUVADIS consortium partners. GEUVADIS FTP site for the scientific data exchange was established at EBI and used successfully by all GEUVADIS partners. Five datasets generated within GEUVADIS consortium have been deposited into EBI archives and are publicly available. GEUVADIS Data Browser was created and successfully used for the analysis results visualization.
3: 500 RNA samples were distributed, and sequenced by seven laboratories, using highlystandardized protocols and quality control measures. The experiments and the core data analysis took place in a highly collaborative manner. Analysis pipelines developed by GEUVADIS partners were used to deliver a high quality analysis: Lappalainen et al., Nature 2013, ‘tHoen et al. Nat Biotech, 2013. Four subgroups were formed and collaborated on medical exome sequencing projects focused on Parkinson’s disease, chronic inflammatory disorders, Fibromyalgia and intellectual disability. We also published and analysed a European-wide survey on existing exome sequencing technologies, tools, use and data storage, access and sharing policies. In addition, we have created a GEUVADIS European Exome Variant Server (GEEVS), hosted at P1, CRG, now accessible to the public at: http://geevs.crg.eu/.
4: We analysed ELSI aspects of phenotype prediction from sequence variation in various clinicalsituations; and in direct-to-consumer (DTC) whole genome sequencing. We also produced a position paper & policy recommendations on ELSI related policy guidelines regarding DTC sequencing. Finally, we performed a study on the analysis of professional attitudes regarding large scale genetic information generated through next generation sequencing in research. During GEUVADIS, we organised/co-organised 11 Training Events and workshops, and co- organised two international conferences.
The project results were disseminated to the scientific community through 176 oral and 22 poster presentations and 43 scientific articles. We also produced a podcast and a video targeting the general public, explaining the main objectives and results of GEUVADIS. The project received important press coverage (23 articles). Overall, the activities performed during the project duration enabled us to design and disseminate standards in operating procedures and biological/medical production and interpretation of RNA and exome sequence data in relation to clinical phenotypes. The consortium brought together cutting-edge knowledge and resources on medical genome sequencing at the European level, and allowed our researchers to develop and test new hypotheses on the genetic basis of disease. We have produced an unprecedented RNA database, as well as an exome variant server, both open to the whole scientific community. Through dissemination, training and ELSI-related activities, our Consortium strongly engaged with society and included all its activities in a wider setting than the restricted genomics research community. Indeed, the rise of sequencing technologies has a strong impact on the daily practice of medicine, on public health systems, and on society in general. Through GEUVADIS, we promoted an efficient, reasonable and responsible use of these technologies in research as well as in clinical practice.
Relevant details of results per work package.
As already mentioned the GEUVADIS Consortium has/ had four main aims, each split in several sub-aims:
- WP1. Coordination and Communication Office. Management and Coordination of the Consortium GEUVADIS and Events (external/ internal).
– Communication and coordination
The CRG (Coordinator Partner; CP) dedicated scientific project management team ensured the daily management and coordination of the project. All project activities were carefully monitored through an efficient communication strategy and through several efficient tools:
– Six specific Mailing Lists, through which the recipient of specific information within one WP (or other specific sub-project group) were targeted.
– Regular Teleconferences, with updates on the project’s activities and discussion on WPs.
– A Website, comprising of a public page, including ‘resources’ sections dedicated to the general public, as well as a podcast explaining the main concept of the project, and a video published by the project “XploreHealth”.
– An intranet accessible to consortium members only, and gathering all useful information on the project; as well as a Wiki – created by the UNIGE team to monitor all activities within WP4
– Three press releases at three project milestone dates: (1) at the kick-off meeting, (2) when the open access to the WP4 generated RNAseq data was given and (3) at the publication of our two collaborative high impact publications.
– Publication of two Interim Reports, prepared and distributed to the SAB before the first and second Annual Meetings. These report enabled monitoring the progress of the project and to get specific feedback from the SAB on how to improve our efficiency within GEUVADIS.
– Events
The Coordinator partner (P1) successfullyorganizedthekick-off(KO) and successive project meetings. Thelist of these events is detailed in table 1 below:
| Type | Date | Comment |
| KO Meeting 2011 | 17/12/2010 | Barcelona, Spain |
| Annual Meeting 2012 | 28/11/2011 | Toulouse, France |
| Annual Meeting 2013 | 29/10/2012 | Santiago de Compostela, Spain |
| Final meeting 2013 | 20/11/2013 | The Hague, the Netherlands |
Table 1
The final meeting was a satellite of the “Hands-on bio banks 2014” event in The Hague, which GEUVADIS co-organized with the BBMRI project, where the general assembly gathered and had an opportunity to discuss the impact of the GEUVADIS project and future activities which could be put in place for the future.
- WP2. Developed Standards in Quality Control (QC) of Sequence Data.
The aim of this WP was to define, establish and disseminate standards for quality control for Next Generation Sequencingof RNA and Exomes, to identify rare mutations.
– Survey
Already established procedures for quality assessment of the sequence data with respect to transcriptome and exome were compared among the Consortium’s participating laboratories. The task was performed by a systematic surveyof the corresponding NGS platforms.
Based on input from all partners, important QC parametershave been defined, listed and are currently applied amongst the partners. A free text survey document has been produced (deliverableQuality control of sequence data), its main conclusion been that the different laboratories, in general, had very similar procedures for sample quantification, target enrichment and sequencing library preparation. However substantial differences were noted in the subsequent data analysis pipelines with respect to filtering procedures, alignment of sequencing reads and variant calling etc.
– Best practice
Best-practice procedures were defined for quality control of transcriptome and exome sequence data from the existing sequencing platforms.
As part of WP4, mRNAs and small RNAs of lymphoblastoid cell lines of 465 individuals were sequenced in a distributed manner by P1-CRG, P2-UNIGE, P3-HMGU, P6-MPIMG, P8-UU, P9- CAU and P11-LUMC. The RNA was extracted by P2-UNIGE and subsequently distributed randomly between the partners who sequenced the samples, following strict guidelines with respect to protocols and version of reagent kits to be used.The success of using this approach has so far manifested itself in two published articles in Nature and Nature Biotechnology.The RNA sequence data produced by the seven centers showed that the biological variation between the individual samples was much larger than the variation between the data from the different labs, thereby demonstrating that distributed RNA-sequencing is feasible.
– Recommendations for the community
These parameters include monitoring: (i) the distribution of nucleotide-level quality scores; (ii) the average and distribution of GC content; (iii) the average and standard deviation of insert size; (iv) the percentage of reads mapping to annotated exons where at least 60% of the mapped reads should map to annotated exons; (v) 3’ to 5’ coverage bias; (vi) and measures should be taken to detect sample swaps, contamination and outliers.
Key quality checks in mRNA and sRNA sequencing are presented in table 2 below:
| Quality checks common to mRNA and sRNA
sequencing |
||
| Distribution of base quality scores | ||
| Average and width of the distribution of GC content | ||
| Percentage of reads mapping to the genome | ||
| Checks for sample swaps and contaminations | ||
| Outlier detection: pair-wise correlations in expression quantification between samples | ||
| Quality checks specific for mRNA | ||
| The average and standard deviation of insert size | ||
| Percentage of reads mapping to annotated exons | ||
| 5′–3′ trends in coverage across transcripts | ||
| Quality checks specific for sRNA | ||
| Length distribution after adaptor clipping | ||
| Percentage of reads mapping to known sRNA genes | ||
Table 2
- WP3. Develop models for Sequencing Data Storage, Access and Exchange.
– Recommendations
Next Generation Sequencing (NGS) data storage and exchange recommendations have been developed and reported. These recommendations consists of three parts:
- MINSEQE (Minimum Information about the Next gEneration Sequencing Experiment)
reporting standard is established and used in ArrayExpress database EBI for NGS data.
- MAGE-TAB submission format is used for NGS data submission and includes a number of recommendations how to describe NGS experiment.
- NGS terms have been added into EFO – Experimental Factor Ontology in order to support qualitative NGS data annotation.
– Analysis components
Analysis components lists have been created and successfully tested internally at EBI and externally by other GEUVADIS consortium members.
The standard analysis components for the variant discovery are:
- Quality control procedures
- Alignment
- Summarization by features procedures
- Normalization procedures
The standard analysis components for the variant discovery are:
- Alignment
- SNP Calling
- Indel Calling
- Structural Variant Discovery
– Data deposition
GEUVADIS FTP site for the scientific data exchange was established at EBI and used successfully by all Geuvadis partners. Data from FTP site have been copied to EBI repositories according to agreements with GEUVADIS RNA-seq project (WP4) partners.
Five datasets generated within the consortium have been deposited into EBI archives and are publicly available:
- GEUVADIS main RNA-seq project mRNA data that passed quality control procedures
(ArrayExpress accession E-GEUV-1),
- GEUVADIS main RNA-seq project small RNA data that passed quality control procedures
(ArrayExpress accession E-GEUV-2),
- GEUVADIS main RNA-seq project all mRNA and small RNA-seq data regardless quality control status (ArrayExpress accession E-GEUV-3),
- GEUVADIS pilot RNA-seq project mRNA data (ArrayExpress accession E-GEUV-4), and
- Dataset “Diagnostic Exome Sequencing in Persons with Severe Intellectual Disability” created by GEUVADIS partner RUNMC (Radboud University, Nijmegen Medical Center) from WP5 is deposited into EGA (“The European Genome-phenome Archive”) under submission EGAS00001000287, dataset’s meta-data are available also through EBI ArrayExpress archive under accession E-GEUV-5.
– Data visualization
GEUVADISdata browser was created especially for the GEUVADIS RNA-seq project to visualize quantification and QTL results and link them together according to the analysis type (transcript, exon,miRNA): http://www.ebi.ac.uk/Tools/geuvadis-das/ Links to the EBI archives with raw and mapped data as well links to the analysis results and genotypes are available from the GEUVADIS data browser.
– EVA (European Variation Archive)
EVA was an EBI new project under development state at the time the GEUVADIS project was up and running. Consortium partners submitted example datasets, participated in conference calls addressed the exome server submission format, and agreed to share the data. EVA plans to follow up with the GEUVADIS project members to initiate a new data submission to the EVA once the submission interface was ready (spring 2014).
- WP4. Handling, Analysis and Interpretation of RNA SequenceData and other Functional Datasets.
The main aim of WP4 was to perform the sequencing and data analysis of 500 RNAseq samples and use the data analysis and eQTL derivation as a means to develop and recommend data production and analysis standard for RNA sequencing.
– Study design
The process of sample preparation, distribution to the 7 sites that will perform the sequencing experiment, organization of data deposition and primary analysis and ultimately the core data analysis towards a publication was by UNIGE through a variety of means, including regular Teleconferences, constant update of the GEUVADIS Wiki site (*) and a continuous collaboration with EBI. A specific mailing list has been created for the analysis group, to target lab members from the GEUVADIS centers who are specifically involved in the sequencing data analysis.
(*) The Wiki site, hosted on the GEUVADIS intranet, is a web-based service which enables both project partnersand external collaborators to enter all information relevant for the project.
Two specific analysis meetings were held in Geneva and Barcelona to agree on analysis plans, and distribute clear tasks to all team members.
– Data analysis
After a collaborative and structured study design in the first half of the project, the following goals were achieved:
- RNAseq data analysiswas completed as part of a coordinated effort among manyeuropean laboratories.
- Raw and processed data were available at public repository for the community to freely use and download (http://www.geuvadis.org/web/geuvadis/RNAseq-project#Data_Access). There is an associated browser (http://www.ebi.ac.uk/Tools/geuvadis-das/) where researchers can visualize both the transcriptome data and the eQTLs discovered in this study.
- Several papers were submitted, two of them published in Nature and Nature Biotechnology. These generated a lot of interest from the community and became a reference for transcriptome studies both in research and medical settings.
– Main scientific findings
The importance of this project lies in three domains:
- The GEUVADIS project shows that the distributed RNA sequencing between seven laboratories has yielded data of extremely high quality, with good replication between different laboratories. Thus, RNA sequencing technology is mature enough for large distributed studies. Furthermore, the shared expertise of the partner laboratories in the most recent methods for e.g. read mapping sets an example for the technical execution of future RNA sequencing studies.
- The dataset that we have created is likely to become one of the most important transcriptome reference datasets for the human genomics community, given the already published genome sequences of these individuals by the “1000 Genomes project”. In addition to making the raw data freely accessible, we are also developing more advanced data sharing and visualization approaches with Ensembl.
- Finally, and most importantly, this project gives us novel insight on how both common and rare genetic variation contributes to quantitative and qualitative transcriptome variation in human populations – not only expression levels, but also e.g. splicing, miRNA-mRNA interactions, fusion genes, and RNA editing. We discover cis-eQTLs for over 8000 genes and alternative splicing QTLs for thousands of genes. The genome sequencing data gives us a more precise view of the spectrum and functional mechanisms of causal regulatory variation. Additionally, analyzing allelic expression and splicing shows how the majority of regulatory variation is rare in the population, thus highlighting the importance of large sample sizes and transcriptome analysis at the level of an individual. Finally, we validate the functional effects of hundreds of loss-of-function variants annotated in these genomes,but also show frequent compensation mechanisms, highlighting how transcriptome sequencing is often essential for proper functional interpretation of genetic variants.
- WP5. Biological and Medical Interpretation of SequenceData for Rare Variants.
– Sequencing tools in Europe
Sequence analysis pipelines have been established at all GEUVADIS partner sites. These pipelines, with few exceptions, use open source software for mapping of sequencing reads to a reference and for subsequent filtering steps to enrich the output variants for properties such as frequency, functionality or pathogenic potential. Further than assessing sequence analysis tools available within the GEUVADIS partners, a much wider survey across Europe was performed, with the aim of collecting comprehensive qualitative and quantitative information on the use of Next Generation Sequencing for research and clinical purposes in Europe. The analysis of the survey results lead to three major conclusions:
- Exome sequencing is a key technology in human genetics research and, to a certain extent, also in clinical genetics
- IlluminaHighSeq 2000, and more generally Illumine, is a key technology in human genomics. A focus on this technology is hence justified in Quality Control and data analysis standardization efforts such as the GEUVADIS WP2 and WP4 RNAseq study.
- Data storage is mostly local and sequencing data is not often shared. This gives a strong justification for the seed to setup user-friendly, secured shared databases of highquality, so that the most is made out of the sequencing data produced throughout Europe. In a context where most sequencing producing institutions are publically funded, this is of especial relevance. Once again, this gives a strong justification for the data storage standardization efforts conducted within GEUVADIS WP3, and for databases such as the GEEVS and the EVA.
– Patient consent and data access
Patient consent issues have been intensely discussed between partner sites (see also WP6 – ELSI). Principles of necessary and eligible points to be addressed in a sequencing consent,were made available by WP6 and appropriate consent forms were established at each partner site.Access to sequencing data is related to the original patient consents, and the interpretation of such consents through investigators and ethics committees.
– Sequencing consortia
Both rare and common variant effect studies in medicine require large samples sizes. Sequencing projects with genome wide data being analyzed in cases and controls, have shown that rare sequence variation using current sequencing technology requires individual level data analyzed centrally. A pilot project within GEUVADIS focused on exome sequences in 500 cases with Parkinson disease cases. Rare variants identified in candidate genes have been performed and will inform the design of customized SNP-arrays. Such array studies in turn will enable large scale association studies testing for both common and rare variants.
– GEEVS: The GEUVADIS European Exome Variant Server
Next Generation Sequencing (NGS) methods have paved the way for in-depth analysis of Mendelian and complex diseases. Identification of rare coding and splicing genetic variants is crucial for the identification of causal genes; hence for effective analysis of clinical samples it is indispensable to distinguish between rare and common variants. As a first step in this direction the 1000 genomes project and the NHLBI Exome Sequencing Project (ESP) have released population allele frequencies for SNPs based on several thousands of samples. However allele frequency (AF) for some SNPs can differ drastically between populations and geographical regions. Unfortunately these region specific alleles will pop up as seemingly rare when comparing variants identified in a sample to the common AF databases. In order to compensate for this issue population/region specific AF databases are required. Here we designed a unified database of human genetic variants in patients of European ancestry. We have collected aggregated SNP frequencies from various European regions, which are provided by the members of the GEUVADIS consortium. The database stores aggregated variant frequencies along with allele counts, and functional annotation based on Refseq, Ensembl and UCSC KnownGenes. We have designed a standard (‘best practice’) protocol for variant calling, quality control and variant aggregation across institutes. The aggregated AFs show a high correlation to 1000genomes and Exome Variant Server AFs. The data is made available via an easy to use web interface ca lled GEUVADIS European Exome Variant Server (GEEVS) or via download at http://www.geuvadis.org/geevs. The website further allows for data submission from any European institute interested in participating in this effort.In December 2013 data from more than1000 cases had been aggregated.
- WP6. Ethical, Legal and Social Issues of Phenotype prediction from Sequence Variation.
Europe has the capacity to play a key role in the integration of sequence data with related phenotypic information, to improve understanding of disease and advance diagnostics and therapy development; this must be implemented in a responsible way and within an harmonized ethics framework that will ensure that these technologies are used for the service of patients and in the respect of fundamental values. Much work had already been done on ELSI in genetics and genomics, thus the work concentrated on the issues that challenge the classical framework through the developing capacities of sequencing, both for research and for translations into the health systems of European countries. They relate to privacy, data protection, results interpretation and communication, and availability of sequence data. In this context GEUVADIS’ first produced documents help in harmonizing the practices in ethical aspects of sequencing within the project through (i) templates for informed consent, (ii) material transfer agreement and, where relevant, (iii) an agreed ethics policy which was set up and transparently communicated through the project’s website. GEUVADIS contributed to the analysis of issues in using high throughput sequencing (1), to the establishment through the European Society of Human Genetics and through the Public health genomics European network of European recommendations for the use of high throughput sequencing in research and clinics (2, 3). GEUVADIS participated in the international debate about return of results and incidental findings in research and in the clinical context in collaboration with other FP7 EU projects or international consortia such as PHGENII, TECHGENE, ESGI, ICGC, EUROGENTEST, 3Gb-TEST, P3G. The views, opinions and self-reflexivity of scientists participating in the project about having their genome sequenced for research and the results were explored and put in a database. In addition to raise awareness and giving the possibility to express the consortium partners’ views, this led to a publication accepted in Journal of Empirical Research on Human Research Ethics. GEUVADIS also contributed to the general reflection concerning the necessity of an international infrastructure for coordinating and improving the efficiency of ELSI analyses in relation to genomics and society (4) and to policy analyses (5). These activities of mapping and coordinating activities in the ELSI domain that are relevant for the issues of phenotype prediction from sequence variation allowed to establish a list of experts and events relevant for the domain as well as proposing training modules, events, resources as well as contributing to policy aspects through answers to public consultations on policy documents. Through the organization of several expert meetings and educational events, GEUVADIS contributed to the update of the community on the evolving data protection framework in the EU, to the establishment of a state of the art on the market of sequencing proposed directly to consumer and to prudent policy aspects in this domain. Finally this coordination action was successful in allowing the scientific and medical community involved to contribute timely to the ethical debates and framework that will allow to keep the patient at the center of technology translation.
Reference:
- Soulier et al.book chapter 2012(deliverable 6.3).
- Van el et al. J HumGenet. 2013 Jun;21(6):580-4.
- Howard et al. Public health genomics, 2013, 16(3):100-9; (deliverable 6.5).
- Kaye et al. Science, 2012, 336, 673-674.
- Meslin et al. ClinTransl Med. 2013 Jul 27;2(1):14.
- WP7. Dissemination and Training
In the past few years, next generation sequencing technologies have become mature and widely adopted, both in research and clinical settings. Still, there are many open questions around data standards, QC, data analysis, and privacy. The GEUVADIS consortium addressed many of these in expert workshops and network meetings.
The GEUVADIS consortium was/ has been very active in dissemination and training. Significant efforts have been placed in increasing the awareness of impact and ethical implications of the use of NGS technologies with the general public. This has been achieved through 3 press releases, 24 general press publications, and 16 oral presentations for the general public. Presentations were mainly focused to educate the general public about NGS technology itself, what people can expect from it, how it will change diagnostics, health care and treatment. The address of ethical concerns, privacy and legal issues received special attention during these lectures.
The introduction of NGS technology in research and in clinical settings raised a hightraining demand. 11 training workshopsfor both basic and clinical researchers and clinicians were organized, as part of an exhaustive training program. These were mainly focused at data analysis routines and (clinical) interpretation of NGS data.
GEUVADIS has also been at the forefront of the ethical and legal discussions associated with NGS technology through the organization of 3 expert workshops with participants from all over the world.
Scientific work and coordinating activities achieved wide exposure in scientific conferences, including conferences on genetics, bioinformatics, medicine, law and bioethics. The GEUVADIS consortium presented a total of 176 scientific talks and 22 posters. Much of the research and discussion has been laid down in a total of 43 scientific publications. The average impact factor of these publications was 12.6, indicative of the high quality, impact and outreach of the work. Uniquely to this project, data associated with the RNA-seq publications (WP4) was already made available to the community in a very early stage, more than half a year before publication in scientific journals. This early access has achieved a spur of further analysis activities by the scientific community, evident from the >300 times the data has been downloaded so far. See: http://www.ebi.ac.uk/Tools/geuvadis-das/ and http://www.geuvadis.org/web/geuvadis/rnaseq-project
Again uniquely to this project, the wiki page in which GEUVADIS researchers archived RNA-seq analysis methods and routines, has been opened up to the community to achieve full transparency and to enhance reproducibility of the results. See: http://geuvadiswiki.crg.es/index.php/Main_Page
Finally, the GEUVADIS European Exome Variant Server (http://geevs.crg.eu/) has been set up (WP5) and opened to the community for the sharing of genetic variants in a secure and privacy- aware manner. This effort has set a standard in the field and increased the visibility of the GEUVADIS consortium.
Impact after dissemination activities of GEUVADIS scientific results.
One of the main scientific achievements of the GEUVADIS project is its RNA study. This study involved RNA sequencing in seven European partner laboratories. In this project, we have sequenced mRNA and micro-RNA of 500 samples from the “1000 Genomes” project, which is thus far the largest study worldwide to combine human transcriptome and genome sequencing data. The project was designed in 2010, the data collected in 2011-2012, and its analysis was completed by end of 2012. Two papers appeared in 2013 (Lappalainen et al. Nature and t’Hoen et al. Nature Biotechnology) and a number of other papers were submitted and accepted afterwards.
The work under this WPwas/is/ will be having a fundamental impact on the way we use transcriptome data for medical studies. We have demonstrated that distributing RNA-sequencing among different laboratories is feasible. This possibility may be particularly attractive for large population- based and cross-biobank studies, where sequencing efficiency and sample logistics may require the combination of data from different laboratories. GEUVADIS has shown that it is feasible to do RNAseq studies in large scale and discover a lot of biological signal. Therefore the value and high information content of the transcriptome has been shown. At GEUVADIS a number of methodologieshave been developed and such a deep biological insight obtainedthat the community will benefit from it in future studies. Given these results it can be anticipated that transcriptome analysis will become a key assay for diagnosis and prognosis in regular medical practice.
These methodologies will also have a higher impact than others produced in smaller scale studies also because of been developed in the context of a consortium which involved the two internationally leading sequencing technologies companies to date, namely Illumina and Life technologies.
This high impact can be illustrated by the article metrics which can be taken directly from the publishers’ websites, as illustrated below for the Lappalainen et al (Nature) and the ‘tHoen et al (NatureBiotec) publications respectively:
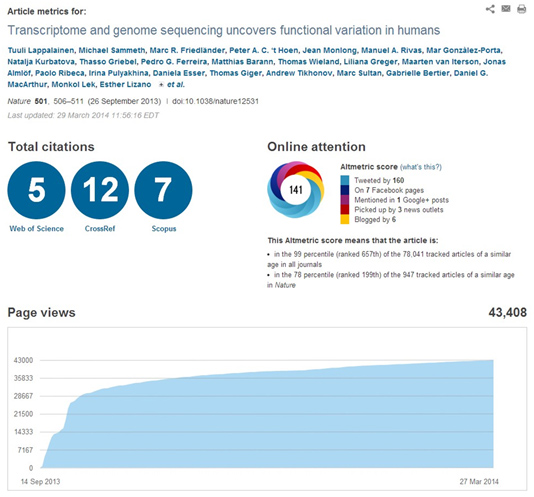
– Impact of Scientific Tools in the Community
The RNA sequencing GEUVADIS study was a success also in the data publicity perspective. Specifically it served as a successful example of a large collaborative project that produced high quality long RNA-seq and small RNA-seq datasets, made the raw (fastq format), processed (bam format) files publicly available, published all protocols and analysis results, created a user friendly analysis results data browser will impact the future of data deposition.
The GEUVADIS data browser has been visited more than 1000 times during the first two months’time of its existence.
GEUVADIS datasets from RNA-seq main project have been downloaded around 1000 times since they were released which make them the most popular RNA-seq studies at EBI ArrayExpressarchive. This result is particularly significant financially; indeed, a large budget is needed by the downloaders to store this data. The total budget needed to store 1000 times the GEUVADIS data largely exceeds the total GEUVADIS budget. Based notably on this simple observation, we claim that wehave managed to generate highly valuable data with a relatively limited funding, complemented by significant investments from the partner institutions.
As described above, we have setup a GEUVADIS European Exome Variant Server (GEEVS). The GEEVS is a unified database of human genetic variants in patients of European ancestry. Aggregated SNP frequencies from various European regions, which are provided by the members of the GEUVADIS consortium. The database stores aggregated variant frequencies along with allele counts, and functional annotation based on Refseq, Ensembl and UCSC KnownGenes. We have designed a standard (‘best practice’) protocol for variant calling, quality control and variant aggregation across institutes. The data is made available via an easy to use web interface called GEUVADIS EuropeanExome Variant Server (GEEVS) or via download at http://www.geuvadis.org/geevs. The website further allows for data submission from any European institute interested in participating in this effort. In December 2013 data from more than 1000 cases had been aggregated. We foresee that this database will be highly useful for the community, and will significantly impact the way exome based clinical studies will be run in the future.
– Impact of the Project on Society
At GEUVADIS we see the adoption of standards for quality control, data analysis, data protection and secure data sharing as the most important GEUVADIS achievement and as essential for the implementation of NGS technology in routine diagnostics and health care. GEUVADIS has played a significant role in the societal debate on the impact and ethical and legal consequences of the introduction of NGS technology, which are essential aspects of its successful implementation. Successful training of biomedical and clinical researchers and clinicians should have helped to ensure the quality of the analysis and interpretation of NGS data.
Indeed, during the course of the project, informative talks have been givenat several local secondary education centers. Numerous open days, in which students were able to see the centers facilities, were also held so the public could come and talk with GEUVADIS researchers about the general objective of the project and it’s potential impact on the future of genomic medicine. GEUVADIS activities were also discussed during a number of lectures for biomedical science and medical students, in addition to the numerous Courses and Workshops organized for students and professionals. Materials for these courses and workshops were always distributed to the participants.
In addition to the dedicated section for the general public in the GEUVADIS website, J. Veltmanwrote a chapter in the book: «Wetenschappelijkedoorbraken de klas in» or “Scientific breakthroughs in the classroom!” by Peeters et al. (ISBN 978-90-818461-1-0).
The podcast we produced on the GEUVADIS project, its objectives and main activities, accessible at http://www.geuvadis.org/web/geuvadis/podcast has been viewed more than 500 times.
The EU project XploreHealth realized a video about GEUVADIS, entitled: ‘do we speak the same genome’ accessible at: http://www.xplorehealth.eu/en/media/do-we-speak-same-genome (more than 9.000 views in December 2013, more than 10.000 views in February 2014). This video was produced by the Reporters Company http://reporters.com.es/. The company won an excellence price for this video: “Premio a unaObraPeriodísticasobreBioética de la FundaciónVíctorGrífolsi Lucas” or the Price to a journalistic work on bioethics from the Victor Grifols y Lucas Foundation. http://www.fundaciogrifols.org/portal/es/2/24857/ctnt/dD7/_/_/8z74/Premios-y-Becas-sobre-Bio%C3%A9tica-2012-2013.html.Since the video is accessible on main platforms such as YouTube and dailymotion, we expect that it will have many viewers will after the project has ended.
– Geuvadis after GEUVADIS / Future Prospects
The QC guidelines we have produced in the context of the RNA sequencing study will be added to the BBMRI web resource, (http://bbmri.eu/) which is updated regularly.
There is also a strong ongoing collaboration between the GEUVADIS RNAsequencing data analysis team with the NIH Genotype-Tissue Expression (GTEx) program, which studies gene expression in human tissues, providing a greatly complementary approach to the one we adopted in GEUVADIS, focusing on human lymphoblastoid cell lines. It is also the largest publically funded RNA sequencing project in the world. A number of GEUVADIS researchers have had a greatly important role in this project by bringing their expertise gathered in the GEUVADIS effort.
Since we are convinced of the potentially strong impact of the GEEVS on the community, we are planning to apply to specific follow-up funding to be able to maintain it at the CRG and extend it significantly by allowing any researcher to submit its data in the server. In addition, we have ensured a strong integration between the EVA (European Variation Archive) which is a fully funded project from the EBI.
Finally, the results obtained in GEUVADIS have also greatly impacted several participating researchers’ careers. Indeed, two postdoctoral researchers, TuuliLappalainen, who drove the RNA sequencing study, as well as Marc Friedlander, who took care of the micro RNA data analysis, managed to obtain an independent positions based in a large part on the results they managed to obtain in GEUVADIS. In addition, several new collaborations in clinical exome sequencing stemmed from the project.
Moreover, several researchers have been called as experts to monitor large scale national and international publically funded large scale sequencing efforts. This is highly significant, since the expertise gathered in GEUVADIS has been recognised by high level decision makers, who are setting the stage for the implementation of clinical genomics at the national, European and international level.
Derived publications
- Transcriptome and genome sequencing uncovers functional variation in humans. Lappalainen et al. Nature, (2013) 501(7468), 506-511. DOI:10.1038/nature12531).
- Reproducibility of high-throughput mRNA and small RNA sequencing across laboratories.‘tHoen et al. Nat Biotech (2013) 31(11), 1015-1022. DOI:10.1038/nbt.2702.
- Veltman; chapter in the book: «Wetenschappelijkedoorbraken de klas in» or “Scientific breakthroughs in the classroom!” by Peeters et al. iSBN 978-90-818461-1-0.

